



La dystonie de l’enfant : »DU DIAGNOSTIC À LA PRISE EN CHARGE »
Nathalie Dorison, Vincent d’Hardemare, Julie Bonheur, Laurent Goetz, Unité DYSPA, Service de neurochirurgie pédiatrique, Hôpital Fondation Rothschild, Paris.
Neurologies • avril 2021 • vol. 24 • numéro 237
| Résumé |
Le terme dystonie désigne un symptôme, mais également un groupe de maladies héréditaires ou acquises. Sa prévalence dans la population générale est estimée entre 15 à 30/100 000/an. Elle est inconnue chez l’enfant. La “paralysie cérébrale” (cerebral palsy) est la principale cause de dystonie de l’enfant, mais les dystonies génétiques sont le plus souvent à début pédiatrique et peuvent mettre en jeu le pronostic vital et fonctionnel de l’enfant. Leur identification et la prise en charge précoce modifient le pronostic à long terme, en particulier si une stimulation cérébrale profonde peut être proposée.
| Définition |
La dystonie est un trouble du mouvement défini comme une « contraction musculaire tonique, involontaire intermittente ou soutenue, entraînant des mouvements répétitifs de torsion et/ou des postures anormales ». Elle provoque des co-contractions de muscles agonistes et antagonistes. Elle peut être initiée ou aggravée par les mouvements volontaires et/ ou des stimuli variés, diffuser vers d’autres muscles (overflow), être diminuée ou disparaître grâce à des gestes antagonistes ou proprioceptifs. Dans le sommeil, nous notons une complète résolution des signes. Elle peut affecter une seule partie du corps (dystonie focale) ou plusieurs régions. Nous parlons de dystonie généralisée lorsqu’elle implique le tronc et au moins deux autres parties du corps.
La dystonie est parfois associée à d’autre types de mouvements anormaux tels que des tremblements, une choréastétose, des myoclonies ou un syndrome parkinsonien.
Lorsque le mouvement anormal prédominant est la dystonie, ce terme désigne la maladie dont l’origine peut être acquise ou héréditaire.
Physiopathologie
Sur le plan physiopathologique, les mécanismes responsables de la dystonie restent encore mal compris à ce jour. La disparité des présentations cliniques et/ou étiologie oblige à envisager des mécanismes physiopathologiques multiples intervenant à différents niveaux du système nerveux central. Plusieurs études chez l’animal et l’humain ont permis de retenir la notion d’une pathologie de réseaux neuronaux (Network disorder) entraînant un dysfonctionnement des boucles de contrôle des fonctions motrices et sensori-motrices « cortex/ganglions de la base/cervelet/tronc cérébral/thalamus » et plus uniquement des ganglions de la base. Parmi les différentes hypothèses évoquées sont retenues une mauvaise intégration sensori-motrice ainsi qu’une désorganisation spatiale et temporelle des activités neuronales impliquées dans le contrôle du mouvement. Elles se traduiraient par une perte d’inhibition de la fonction motrice « Surround inhibition » et conduirait à l’activation désorganisée (contraction) et excessive de groupes musculaires. [1]
Orientation diagnostique devant une dystonie de l’enfant
Classifications et causes génétiques principales
En 2013, Albanese a proposé une classification des dystonies fondée sur la clinique et l’étiologie (Tab. 1) [2]. L’interrogatoire, l’examen neurologique avec analyse sémiologique des mouvements anormaux, l’examen somatique global et le plus souvent une IRM cérébrale sont les éléments indispensables pour s’orienter devant un patient dystonique. Nous distinguons schématiquement les dystonies héréditaires (d’origine génétique supposée ou confirmée), et secondaires (acquises fixées ou progressives, ces dernières pouvant cependant être d’origine génétique…). Le bilan étiologique sera orienté en fonction de cette classification.
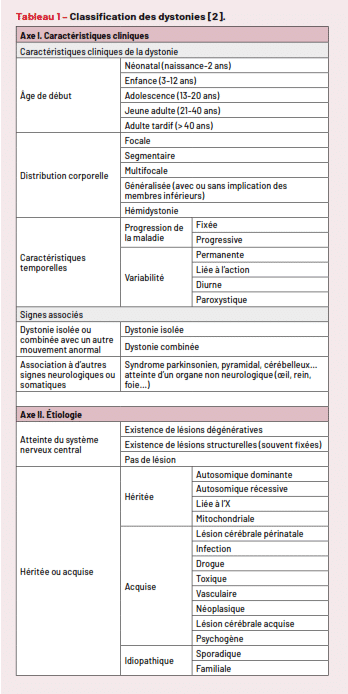
Tableau 1 – Classification des dystonies
Dystonies primaires
Pas d’autres mouvements anormaux associés (en dehors du tremblement), pas d’autre signe neurologique ; les examens d’imagerie (IRM) sont normaux. Elles sont d’origine génétique supposée ou prouvée, avec souvent une hétérogénéité familiale et une pénétrance variable. Elles apparaissent le plus souvent dans l’enfance et ont tendance à la généralisation. La plus connue est la DYT1 (dystonie musculaire déformante).
Dystonies primaires « plus »
D’autres mouvements anormaux (myoclonies, mouvements choréiques, syndrome parkinsonien…), des troubles cognitifs et des atteintes extr
neurologiques peuvent être présents. En font partie les dystonies dopasensibles et la dystonie myoclonique (DYT11). Des anomalies dans le gène KMT2B semblent être l’une des causes principales de dystonie progressive de l’enfant (dystonie généralisée avec atteinte cervico-faciale et aphonie progressive chez l’enfant, en association à d’autres mouvements anormaux et à des troubles cognitifs inconstants) [3].
Dystonies et dyskinésies paroxystiques
D’origine génétique (par ex-mutation dans ADCY5).
Dystonies secondaires acquises « fixées »
Séquelles de prématurité, d’AVC, ictère nucléaire, traumatisme, infection… Les IRM montrent des anomalies typiques, l’atteinte n’est pas progressive. Ce sont les plus fréquentes chez l’enfant.
Dystonies secondaires progressives sur maladie hérédodégénérative et/ou métaboliques
PKAN, acidurie glutarique, Lesch Nyhan, Wilson, Huntington, maladies mitochondriales leucodystrophies, SEP…
Dystonies post-médicamenteuses (en particulier post-neuroleptiques) Rares chez l’enfant.
Dystonies fonctionnelles
Doivent amener à rechercher des abus sexuels, dont la fréquence est sous-estimée. Elles sont exceptionnelles avant 10 ans.
Particularités cliniques de l’enfant
Chez l’enfant la présentation clinique présente quelques particularités cliniques (Tab. 2).
 Tableau 2 – Particularités cliniques de l’enfant
Tableau 2 – Particularités cliniques de l’enfant
L’interrogatoire recherchera particulièrement les points suivants :
- notion d’antécédents familiaux et périnataux ; dans les atteintes génétiques, il existe une hétérogénéité clinique et une pénétrance variable qui peut compliquer l’interrogatoire. Par ailleurs, une souffrance périnatale n’exclut pas une atteinte génétique associée (cf infra) ;
- présence d’une hypotonie axiale avec hypertonie des membres chez le nourrisson qui est souvent le premier signe ; l’apparition des mouvements anormaux et de l’hypertonie axiale est secondaire ;
- sensibilité au jeûne, à l’effort, et aux infections orientant vers certaines atteintes métaboliques, en particulier liées au glucose ou à la mitochondrie.
La reconnaissance du mouvement anormal prédominant chez l’enfant est parfois difficile, en particulier dans un contexte de lésion cérébrale ou de mouvements paroxystiques, en raison de l’association de différents types de mouvements anormaux et de leur fluctuation au cours d’une journée. Les vidéos familiales peuvent être d’une grande aide pour l’analyse. La dystonie peut débuter brutalement, en particulier dans les dystonies génétiques, avec un trigger d’allure parfois psychologique (stress, peur, chute minime), qui peut amener à de faux diagnostic de trouble fonctionnels.
L’analyse des dossiers en réunion de concertation pluridisciplinaire (RCP) après accord des parents permet d’augmenter le rendement diagnostic (diagnostic chez 34 % de patients restés sans étiologie pour Van Egmond et al.) [4].

Figure 1 Patiente avec dystonie cervicale sur mutation KMT2B.
Modification du schéma corporel : copie d’un modèle sans inclinaison de la tête.
Examens à visée étiologique
> Examens radiologiques
- L’IRM cérébrale (avec au minimum séquences T1, T2, T2*, Flair, SWI) reste l’examen de première intention, mais nécessite souvent une anesthésie générale ou une sédation dans ce contexte de mouvements anormaux. Elle recherche des anomalies des noyaux gris pouvant orienter le diagnostic ; une spectroscopie est utile en cas de suspicion de maladie métabolique.
- Le scanner cérébral peut être intéressant à la recherche de calcifications.
> Examens biologiques
Ils seront orientés en fonction de l’anamnèse, de l’examen clinique et de l’IRM. Ils comportent le plus souvent un bilan sanguin à visée métabolique, une ponction lombaire et un bilan urinaire.
> Examens génétiques
Des études par panel de gènes, exomes en trio, voire analyse du génome, permettent de confirmer une étiologie génétique dans de nombreux cas, en particulier de dystonie généralisée ou à début précoce, très fréquente chez l’enfant. Cependant, le délai de rendu des analyses génétiques justifie à l’heure actuelle la poursuite des examens radiologiques et métaboliques afin de ne pas méconnaître une pathologie curable (cf infra).
« Pièges” diagnostiques en pédiatrie
Causes traitables de dystonie
Elles sont rares, mais leur dépistage rapide est indispensable afin de limiter les conséquences parfois irréversibles des lésions qu’elles provoquent (Tab. 3).
Toute dystonie fluctuante justifie en particulier une ponction lombaire pour analyse des neurotransmetteurs et dosage de la glycorachie (associée à une glycémie dans le même temps) afin de ne pas méconnaître un déficit en transporteur de glucose (Glut1) ou une dystonie dopasensible (maladie de Segawa et apparentées) qui peut se présenter comme une dystonie isolée, un syndrome akinétorigide ou un tableau de « pseudo paralysie cérébrale ». Le dépistage néonatal de l’acidurie glutarique par spectrométrie de masse est en cours de généralisation en France depuis 2020, mais n’est pas encore généralisé à tout le territoire.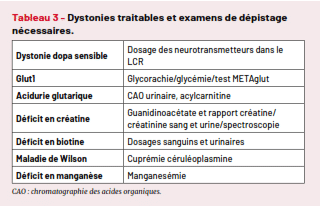
Tableau 3 – Dystonies traitables et examens de dépistage
« Pseudo » paralysie cérébrale (PC)
La prévalence de l’encéphalopathie anoxo-ischémique néonatale est estimée entre 1 et 25/1 000 nouveau-nés vivants ; 15 à 20 % d’entre eux vont évoluer vers une paralysie cérébrale ; les mouvements anormaux incluant la dystonie sont estimés à 15 % des cas. Ce diagnostic est cependant surestimé et doit être retenu sur des critères cliniques et paracliniques rigoureux, en particulier à l’IRM. Environ un tiers des patients ayant un diagnostic de PC dyskinétique auraient une IRM normale et une pathologie génétique sous-jacente ; il faut penser particulièrement aux dystonies dopasensibles et aux mutations ADCY5 [5]. Par ailleurs, les nouveau-nés ayant une pathologie neurologique constitutionnelle sont plus à risque de présenter une anoxo-ischémie néonatale, mais leur évolution sera atypique (dégradation secondaire, modification sémiologique). Une IRM est indispensable pour confirmer le diagnostic de PC et est à renouveler ± associée à un bilan métabolique et/ou génétique en cas d’évolution atypique.
L’encéphalopathie hyperbilirubinémique (ou ictère nucléaire)
La dystonie est souvent sévère, fixée, associée à des mouvements choréoathétosiques ; la surdité est fréquente, le développement cognitif est relativement préservé. L’IRM cérébrale peut montrer des hypersignaux T2 et FLAIR des globes pâles.
Le risque semble accru chez les grands prématurés, malgré la photothérapie, les taux toxiques variant en fonction de l’état nutritionnel (hypoalbuminémie) et infectieux et étant mal connus. Une sortie précoce de maternité ou un accouchement à domicile « volontaire » sans surveillance clinique sont retrouvés dans l’anamnèse de certains cas.
La dystonie fonctionnelle ou psychogène
La prévalence des mouvements anormaux psychogènes chez l’enfant n’est pas connue, mais le diagnostic est retenu pour 3 % des enfants consultant pour mouvements anormaux ; le plus souvent il s’agit de tremblements, dystonie ou troubles de l’équilibre. Exceptionnel lorsque les signes débutent avant 10 ans, ce diagnostic doit rester un diagnostic d’élimination en pédiatrie. Comme chez l’adulte, dans plus de 75 % des cas, les manifestations surviennent chez des filles, avec une surreprésentation de traits perfectionnistes et de syndromes anxiodépressifs. Une histoire d’abus sexuel est retrouvée dans 6 % des cas (sans doute sous-estimée). Les signes d’appel sont le plus souvent : une dystonie douloureuse avec des troubles sensitifs non anatomiques et/ou une sensation de faiblesse musculaire,avec un cortège de signes fonctionnels variés. Le critères cliniques suivants sont souvent pris à tort pour des critères d’allure psychogène et sont au contraire en faveur d’une étiologie organique: association de divers types de mouvements, rémission spontanée, dystonie induite par une tâche spécifique ou l’action, existence d’un geste antagoniste, aggravation ou apparition après un stress ou une émotion, épisodes paroxystiques ou variations diurnes [6].
Traitements de la dystonie
Traitement médical (Tab.4)
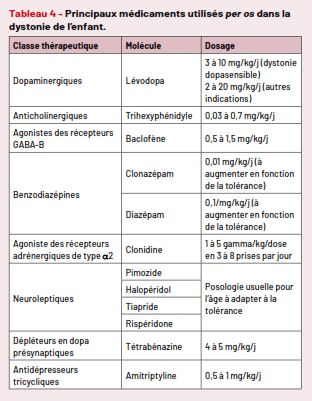
Tableau 4 – Principaux médicaments utilisés per os dans la dystonie de l’enfant
Les traitements sont prescrits le plus souvent hors AMM et les doses sont calculées en fonction du poids. Les galéniques sont souvent inadaptées pour le petit enfant, nécessitant des reconditionnements ou des manipulations parfois délicates pour les parents. Leur efficacité est partielle, mais ces traitements sont d’autant plus efficaces qu’ils sont instaurés avant l’apparition des déformations orthopédiques. L’augmentation des posologies se fait lentement, pour améliorer la tolérance ; le sevrage rapide peut être source d’aggravation des signes. La plupart des médicaments ont un retentissement sur la mémoire et la vigilance ; ils peuvent également accroître le bavage et les troubles de déglutition déjà fréquents dans cette population. L’efficacité, l’indication et la tolérance des traitements nécessitent une réévaluation régulière par le médecin prescripteur. Nous ne parlerons pas ici des traitements spécifiques des maladies métaboliques ou traitables, mais nous retiendrons qu’un test thérapeutique par 3 mois de dopa est conseillé devant toute dystonie inexpliquée de l’enfant afin de ne pas méconnaître une dystonie dopasensible.
Les douleurs sont souvent sous- estimées par les médecins et les rééducateurs et peu ou non verbalisées spontanément par les patients. Une attention particulière doit y être portée, car elles sont source d’aggravation de la dystonie (« épine irritative »). Il peut s’agir de douleurs liées à la dystonie, mais également de douleurs liées à l’appareillage, à un reflux gastrique, une ostéoporose (fréquente chez le jeune polyhandicapé), à des causes orthopédiques, des douleurs dentaires, etc. [7, 8].
Traitement de l’état de mal dystonique (status dystonicus, orage dystonique)
Il s’agit d’une urgence pouvant engager le pronostic vital. Il est caractérisé par des accès dystoniques répétés ; nous distinguons la forme tonique (contractions et postures anormales soutenues) de la forme phasique (contractions rapides et répétitives). La forme tonique est de moins bon pronostic et survient plus fréquemment chez le garçon et dans les dystonies acquises secondaires. Le plus souvent le status survient après une phase d’aggravation de la dystonie. Le risque est l’évolution vers un dysfonctionnement bulbaire, la défaillance respiratoire et une rhabdomyolyse avec défaillance multiviscérale. Il survient dans 60 % des cas entre 5 et 16 ans, quelle que soit la cause de la dystonie [9].
Lumsden et al. ont établi un plan d’action en cas de décompensation de la dystonie pour éviter l’évolution vers un état de mal [10].
Dans deux tiers des cas on retrouve un facteur favorisant (Tab. 5). Le traitement est basé sur l’antalgie, l’hydratation, la sédation, allant jusqu’à l’intubation ventilation assistée. La stimulation cérébrale profonde en urgence doit être discutée rapidement si le status ne cède pas aux thérapeutiques médicamenteuses.

Tableau 5 – Facteurs favorisants les états de mal dystoniques (trigger)
Traitement neurochirurgical : la stimulation cérébrale profonde (Deep Brain Stimulation)
Cette intervention est proposée à certains enfants dystoniques (cf infra).
La stimulation cérébrale profonde (SCP) est une approche thérapeutique médico-chirurgicale permettant de stimuler électriquement une structure profonde du cerveau impliquée dans le contrôle du mouvement. Dans le cadre des dystonies, la cible la plus fréquente est le globus pallidus interne (Fig. 2).
Technique opératoire de la stimulation cérébrale profonde
La chirurgie d’implantation stéréotaxique d’électrode se déroule sous anesthésie générale avec un contrôle per- opératoire d’imagerie et d’électrophysiologie pour affiner la position de l’électrode dans la partie postéro-ventrale du globus pallidus interne. Dans les dystonies secondaires notamment dégénératives, d’autres cibles ou des schémas à plusieurs électrodes sont en cours d’étude et peuvent être retenus. Dans la seconde partie de la chirurgie, les électrodes sont connectées à un générateur de pulse délivrant une stimulation électrique à haute fréquence (130 Hz) au niveau de la structure cible. Ce générateur est placé en position abdominale ou pectorale et relié aux électrodes par des connecteurs et des extensions sous-cutanées. Il est à changer tous les 3 à 10 ans en moyenne selon le modèle choisi (non rechargeable ou rechargeable) (Fig. 2b).
Complications de la SCP
La SCP est une méthode de traitement réversible, les complications opératoires, en particulier hémorragiques ou ischémiques, sont rares et le risque essentiel reste l’infection du matériel dans 3 à 10 % des cas qui nécessite son ablation dans 85 % des cas. Cette infection peut également survenir à long terme comme pour tout porteur de matériel étranger, à l’occasion d’une infection d’origine dentaire orthopédique ou digestive le plus souvent. Ce risque semble plus important en cas de dénutrition et de polyhandicap. Des dysfonctions ou fractures des extensions ou des électrodes peuvent survenir, en particulier après plusieurs années, et nécessiter une reprise au bloc opératoire (18 % des cas). Une dégradation transitoire de la dystonie est fréquente en post-opératoire immédiat (facteurs douloureux et émotionnels le plus souvent). Un repositionnement des électrodes ou un ajout d’électrodes peut être nécessaire chez certains patients, en particulier ceux opérés jeunes [11].
Résultats généraux de la SCP dans la dystonie
Dans les dystonies primaires, elle peut apporter une diminution de 70 à plus de 90 % des mouvements anormaux.
Les résultats sont beaucoup plus aléatoires dans les dystonies secondaires, avec un bénéfice très variable, mais en général inférieur à 30 % sur la composante motrice. Ces résultats reflètent la difficulté à recréer des réseaux fonctionnels sur un cerveau lésé et également à repérer les « bons candidats » pour cette stimulation cérébrale profonde (SCP).
Les effets de la SCP dans la dystonie portent surtout sur les formes mobiles ou tremblantes et les accès aigus douloureux, ainsi que les « status dystoniques ». Il peut y avoir une amélioration de la déglutition, de la dysarthrie et de la protrusion dystonique de langue. La SCP est en général inefficace sur les postures dystoniques fixées, les dystonies très focales des membres, les troubles de l’équilibre et de la posture.
Il n’y a pas d’effet sur l’hypotonie et la cognition. Nous notons cependant une amélioration cognitive chez certains patients, sans doute en rapport avec la diminution des traitements et l’amélioration de la motricité [12].
> Résultats de la SCP dans les dystonies secondaires
Les “bonnes” indications de la SCP dans les dystonies secondaires ne sont pas encore clairement établies.
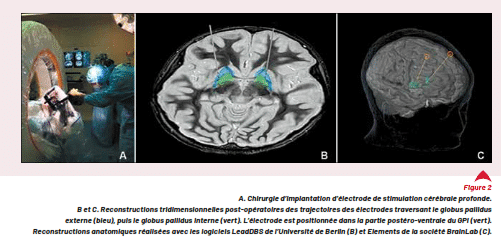
Figure 2
A. Chirurgie d’implantation d’électrode de stimulation cérébrale profonde.
B et C. Reconstructions tridimensionnelles post-opératoires des trajectoires des électrodes traversant le globus pallidus externe (bleu),
puis le globus pallidus interne (vert). L’électrode est positionnée dans la partie postéro-ventrale du GPi (vert).
Reconstructions anatomiques réalisées avec les logiciels LeadDBS de l’Université de Berlin (B) et Elements de la société BrainLab (C).
Paralysie cérébrale dyskinétique
La SCP peut être envisagée pour diminuer en particulier les accès dystoniques, avec un moindre effet à attendre sur les dyskinésies. Le gain fonctionnel est limité, mais les résultats sont très hétérogènes dans cette population. La plupart des patients rapportent un gain sur les contractures, les douleurs, une plus grande facilité d’installation et une meilleure tolérance de l’appareillage ; la qualité de vie et le confort sont améliorés. Les données pédiatriques sont rares, mais l’amélioration semble possible au-delà de 1 an post-implantation et l’effet se maintient au-delà de 2 ans. L’évaluation clinique de ces patients est difficile et incomplète, les échelles disponibles pour évaluer l’efficacité de la SCP (en particulier la BFMDRS) ayant été développées pour les dystonies primaires et n’étant pas assez sensibles pour cette population [13].
Maladies métaboliques ou hérédodégéneratives
La SCP peut également être proposée à des patients ayant des dystonies secondaires, en rapport avec des maladies métaboliques ou hérédodégéneratives, comme les NBIA (neurodégénérescence avec surcharge cérébrale en fer), en particulier celles liées aux mutations PANK2 ou « PKnopathie » (PKAN) et les patients présentant des automutilations sur syndrome de Lesh Nyhan. Les aciduries glutariques sont également une indication discutable chez l’enfant, en particulier pour diminuer les accès aigus douloureux et les dyskinésies.
Information des patients et des familles à propos des résultats de la SCP
Il y a peu de moyens pronostiques à l’heure actuelle pour évaluer les bons candidats à la SCP en particulier dans les dystonies secondaires. Les effets attendus seront fonction de la maladie sous-jacente et l’amélioration sera partielle, avec un échappement après quelques mois ou années pour les maladies hérédodégénératives. Le gain fonctionnel n’est souvent pas le but principal de la SCP dans cette population. Ces données doivent être clairement exposées aux patients et familles en préopératoire et le rapport risque/ bénéfice de l’intervention, évalué de façon pluridisciplinaire. L’équipe de J.P. Lin [14] a été la première à souligner l’importance d’intervenir le plus précocement possible par rapport au début de la dystonie, et de fixer des objectifs personnalisés réalistes en préopératoire, afin de ne pas aller au-devant d’une déception. Ils se sont intéressés au mécanisme de décision parentale. Un point particulièrement intéressant est que les parents semblent ressentir un sentiment d’avoir « plus à perdre » en cas d’échec de l’intervention lorsque le handicap est modéré, ce qui peut influer leur décision et retarder l’âge de prise en charge.
Toxines botuliques
Utilisées comme traitement de choix dans les dystonies focales ou segmentaires prédominantes chez l’adulte, elles n’interviendront qu’en appoint dans la plupart des dystonies de l’enfant qui ont tendance à être généralisées ou multifocales. Elles seront discutées en particulier pour les dystonies à prédominance cervicales, moins accessibles aux traitements médicamenteux ou à la SCP, ou au niveau des membres pour lutter contre une déformation ou une douleur localisée. Elles nécessitent des injections répétées tous les 3 à 4 mois en général, avec une dose maximale par patient et par séance.
Pour les patients avec paralysie cérébrale, elles sont utilisées pour lutter contre la spasticité.
Correspondance :
Nathalie Dorison Unité DYSPA, service de neurochirurgie pédiatrique, Hôpital Fondation Rothschild,
29 rue Manin, 75019 Paris
✖ Nathalie Dorison déclare avoir des liens d’intérêt avec PTC, Medtronic et Boston.
✖ Julie Bonheur déclare avoir des liens d’intérêt avec Boston.
✖ Vincent d’Hardemare déclare avoir des liens d’intérêt avec Boston, Medtronic et Abbott
Bibliographie
- Balint B, Mencacci NE, Valente EM et al. Dystonia. Nat Rev Dis Primers 2018 ; 4 : 25.
- Albanese A, Bhatia K, Bressman SB et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord 2013 ; 28 : 863-73.
- Cif L, Demailly D, Lin JP et al. KMT2B-related disorders: expansion of the phenotypic spectrum and long-term efficacy of deep brain stimulation. Brain 2020 ; 143 : 3242-61.
- van Egmond ME, Eggink H, Kuiper A et al. Crossing barriers: a multidisciplinary approach to children and adults with young-onset movement disorders. J Clin Mov Disord 2018 ; 5 : 3.
- Pearson TS, Pons R, Ghaoui R et al. Genetic mimics of cerebral palsy. Mov Disord 2019 ; 34 : 625-36.
- Ferrara J, Jankovic J. Psychogenic movement disorders in children. Mov Disord 2008 ; 23 : 1875-81.
- Lumsden DE, Kaminska M, Tomlin S et al. Medication use in childhood dystonia. Eur J Paediatr Neurol 2016 ; 20 : 625-9.
- Roubertie A, Mariani LL, Fernandez-Alvarez E et al. Treatment for dystonia in childhood. Eur J Neurol 2012 ; 19 : 1292-9
- Allen NM, Lin JP, Lynch T, King MD. Status dystonicus: a practice guide. Dev Med Child Neurol 2014 ; 56 : 105-12.
- Lumsden DE, King MD, Allen NM. Status dystonicus in childhood. Curr Opin Pediatr 2017 ; 29 : 674-82.
- Kaminska M, Perides S, Lumsden D et al. Complications of Deep Brain Stimulation (DBS) for dystonia in children—the challenges and 10 years experience in a large paediatric cohort. Eur J Paediatr Neurol 2017 ; 21 : 168–75.
- Badhiwala JH, Karmur B, Elkaim LM, etal. Clinical phenotypes associated with outcomes following deep brain stimulation for childhood dystonia. J Neurosurg Pediatr 2019 : 1-9.
- Koy A, Timmermann L. Deep brain stimulation in cerebral palsy: Challenges and opportunities. Eur J Paediatr Neurol 2017 ; 21 : 118-21.
- Lumsden DE, Kaminska M, Gimeno H et al. Proportion of life lived with dystonia inversely correlates with response to pallidal deep brain stimulation in both primary and secondary childhood dystonia. Dev Med Child Neurol 2013 ; 55 : 567e74.
 En cours de chargement…
En cours de chargement…





